Introduction¶
Author: JL Villanueva (joseluis.villanueva@crg.eu)
This report describes the differential gene expression analysis comparing WT and sh samples (NUDIX5). We will use Kallisto (v 0.43.0) for quantification at the transcript level and Sleuth (v 0.30.0) for testing differential expression (DE) aggregating transcripts into genes.
Initial samples¶
| num | sample | condition | sequencing |
|---|---|---|---|
| 1 | rw_026_01_01_rnaseq | 2D_wt | 06/27/2018 |
| 2 | rw_030_01_01_rnaseq | 2D_wt | 06/27/2018 |
| 3 | rw_031_01_01_rnaseq | 2D_sh | 06/27/2018 |
| 4 | rw_032_01_01_rnaseq | 2D_sh | 06/27/2018 |
| 5 | rw_039_01_01_rnaseq | 3D_wt | 07/24/2018 |
| 6 | rw_039_02_01_rnaseq | 3D_wt | 07/24/2018 |
| 7 | rw_040_01_01_rnaseq | 3D_sh | 07/24/2018 |
| 8 | rw_040_02_01_rnaseq | 3D_sh | 07/24/2018 |
Mapping statistics¶
| X | sample | reads_mapped | reads_proc | frac_mapped | bootstraps_present | bootstraps_used | condition |
|---|---|---|---|---|---|---|---|
| 1 | rw_026_01_01_rnaseq | 44375965 | 47925975 | 0.9259 | 100 | 100 | 2D_wt |
| 2 | rw_030_01_01_rnaseq | 55177695 | 59107148 | 0.9335 | 100 | 100 | 2D_wt |
| 3 | rw_031_01_01_rnaseq | 44576517 | 48206868 | 0.9247 | 100 | 100 | 2D_sh |
| 4 | rw_032_01_01_rnaseq | 56474041 | 61169209 | 0.9232 | 100 | 100 | 2D_sh |
| 5 | rw_039_01_01_rnaseq | 52429267 | 57390135 | 0.9136 | 100 | 100 | 3D_wt |
| 6 | rw_039_02_01_rnaseq | 41912042 | 44742750 | 0.9367 | 100 | 100 | 3D_wt |
| 7 | rw_040_01_01_rnaseq | 47846169 | 51477759 | 0.9295 | 100 | 100 | 3D_sh |
| 8 | rw_040_02_01_rnaseq | 47094242 | 50976488 | 0.9238 | 100 | 100 | 3D_sh |
Considerations¶
Initially we wanted to test wt vs sh in 2D and 3D and also 2Dwt vs 3Dwt / 2Dsh vs 3Dsh. However there is a batch effect as can be seen in the sequencing date, making it impossible to do the latter comparison.
Therefore we will only test wt vs sh (2D and 3D).
Code for the whole analysis is available in scripts folder. Figures in Figures and tables in tables folders respectively.
PCA With all samples¶
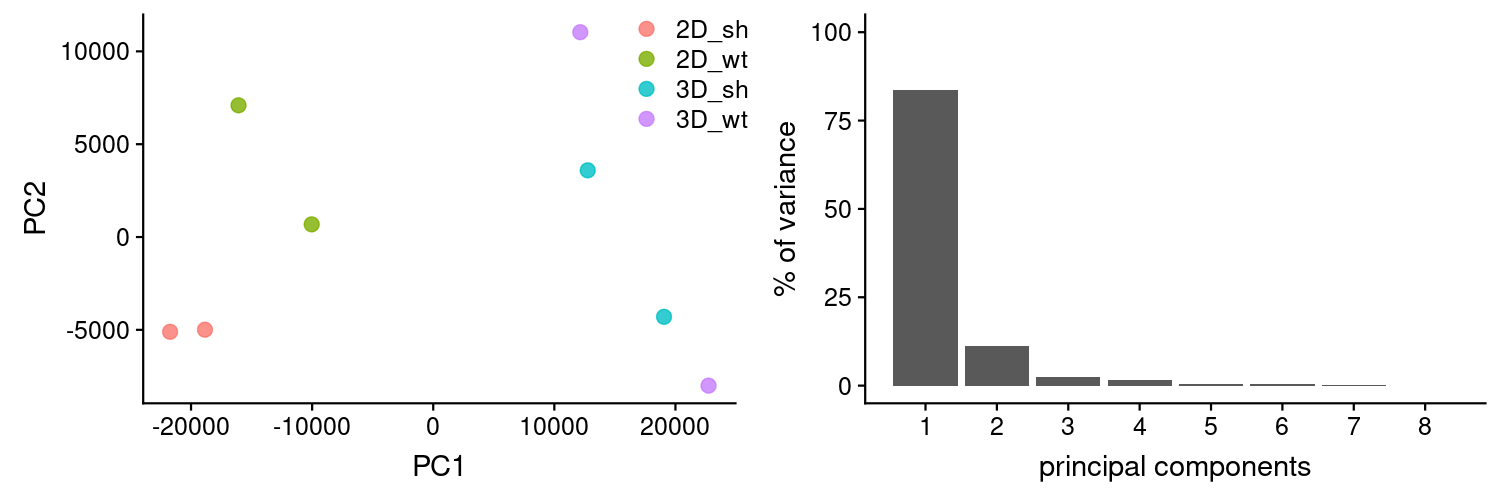
PCAs were generated using TPMs as calculated by Kallisto. It looks like one of the replicas from 3D_wt might have been swapped with 3D_sh. We will explore this later on.
2D_wt vs 2D_sh¶
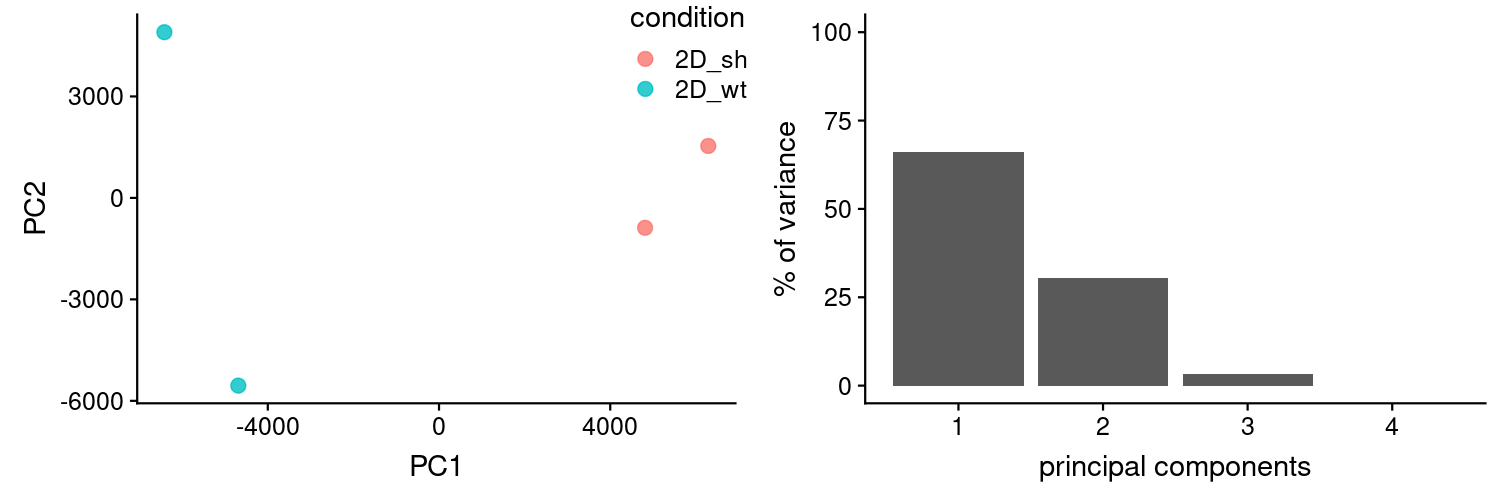
PCA looks good.
Summary table with mean counts (per condition) per gene is named: sleuth_table_wTPM_2D_wt_vs_sh.csv
DE analysis¶
We do the DE analysis aggregating transcripts into genes. We use the likelihood ratio test (lrt). For this comparison we obtain 350 genes.
Summary table for significant genes is named significant2D_wt_vs_sh.csv
Because for this test sleuth does not return a fold change or equivalent metric, I have calculated 3 additional columns: tpm_wt and tpm_sh that are the sum of the average tpm (in replicas) for all transcripts that belong to a gene. After that we have tpm_wt_by_sh, that simply divides log2(tpm_wt +0.1/ tpm_sh+0.1) with a pseudocount of 0.1. Therefore we have a relative measure of the change between wt and sh.

An example of a DE gene: NUDT5¶

3D_wt vs 3D_sh¶
Summary table with mean counts (per condition) per gene is named: sleuth_table_wTPM_3D_wt_vs_sh.csv
We checked the patterns of ALDH1, NUDT5, OCT4 (POU5F1), CD44 that are supposed to be upregulated in wt. NUDT5 gene has a profile in the samples compatible with the sample tags shown in the PCA. The other genes can't be found (ALDH1) or have inconsistent patterns across transcripts. Therefore we perform the DE analysis using the original tags although the clustering in the PCA is not very good.
We get 3 DE genes using lrt test.
Summary table for significant genes is named significant3D_wt_vs_sh.csv